Developing a molecule database is not a trivial task. There are several problems you have to deal with and they can be handled differently depending on the size, content and purpose of the database. Here I try to provide a list of the most common issues we came across recently. First: multiple components.
Part 1. Multiple components.
It usually makes sense to handle single components separately even if samples are provided as multiple components in the source data format. This enables structure searching/filtering at the level of single components. Also, you always have to be suspicious with multiple component entries. The sample might be a mixture of individual, equally important components, but it is also possible that the components are in a particular relationship. It is crucial to analyze this relationship before registering any of the components. Entries coming from vendor companies might contain the followings: salt counter ions, solvent molecules, additives (e.g. antioxidants), contaminants, intermediers and different stereoisomers. Some interesting examples you can see here:

It is essential to determine what the components are and what the relationship between them is. In several cases, however, this is not obvious. Problems arising from these discrepancies are (among others): assigning correct purity (depends on whether additional components are counter ions or contaminants), assigning correct stereochemistry (are the components stereoisomers of each other?), recognizing incorrect drawing (e.g. missing bonds), etc.
In fact, even industry standard tools handle these issues superficially. One widely used approach is to retain the largest fragment (larger MolWeight or nb of heavy atoms) and discard the remaining components of the entry. This can lead to several problems, all arising from loosing important information (all components were there for a reason…).
Identifying salts
Even if we assume that only the main component and counter ion(s) can get into our entries, we will fail to identify several complex organic counter ions when retaining the largest fragment only:

One possible, but rather time-consuming approach (mcule is following at the moment) is to build a salt knowledge base. During registration we check for these frequently used counter ions automatically. This can also work for solvents and typical additives, but it has to be kept in mind that these molecules can also be the main components of some entries.
The picture gets even more complicated when accounting for counter ions represented as covalently bound to the main component. One approach that partially handles this situation is InChI. InChI disconnects all metals from the molecule, and when it is called with the -RecMet option (Note: This will result in a non-standard InChI!) those metals single bonded to some sort of acids (considered as salt counter ions) will not be reconnected. InChI definitely works in some cases, but due to its incomplete acid definitions it fails to identify some important salts, e.g.:

ChemAxon's Structure Checker also have this function. And it seems to successfully identify the counter ions of the examples above. I haven't tested it extensively yet though.
Identifying intermediers
To find out whether a vendor drew the main component together with an intermedier, is another challenging task. Intermediers are usually very similar to the main component.

In some cases, the intermedier is exactly the substructure of the main component. This can be easily checked by a substructure search.

It is also common, however, that the intermedier contains slightly different end groups compared to the perfectly matching substructure. A fingerprint-based similarity search might reveal an intermedier. The metric used here, however is critical. Instead of the Tanimoto coefficient, you might use the asymmetric Tversky index by focusing on the structural elements of the smaller intermedier only. These searches can get an initial guess, but suspicious cases should be checked manually.
Identifying stereoisomers

Due to the ambiguities in the Mol file specification, stereoisomers can be represented in several ways in Mol files (I will write about these in detail in another post). One possibility to represent stereoisomers in a sample is to draw them as multiple components. In this case, comparing the InChI strings of the individual components should reveal that they are stereoisomers indeed. Their InChIs should be identical if the stereo layers are omitted. Linear fingerprints of stereoisomers should be identical too.
Identifying duplicates
For stochiometric reasons, exact duplicates might exists in a single entry. These can be identified by an exact InChI match. Fingerprints will also find these molecules identical.

Identifying drawing problems
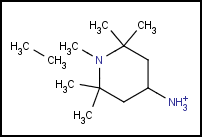
One further reason for multiple fragments might be a missing bond between two parts of the same molecule. Analysis of the 2D coordinates might give an indication for such a drawing error, but proximity of two atoms does not necessary mean they were meant to be connected. Unfortunately, the Mol representation does not help in recognizing these errors from valence states. The absence of a bond typically does not cause valence errors - the missing valences are assumed to be filled by implicit hydrogens.
--
Comments on the above problems or others not mentioned in this post are welcome! Solutions (free or commercial) are highly appreciated too!
No comments:
Post a Comment